Nanomatch Morphology Calculator
Properties |
Notes |
|---|---|
byproduct |
|
Dipole (property not yet described / available) |
byproduct |
recommended |
Workflow
The Nanomatch Morphology Calculator workflow is essentially the first part of the Nanomatch Mobility Calculator workflow (Nanomatch Mobility Calculator), which is terminated after the morphology is generated and analyzed.
Nanomatch Software |
Scientific Role |
Illustration |
|---|---|---|
Geometry optimization
|

|
|
Computation of intramolecular
forcefields
|
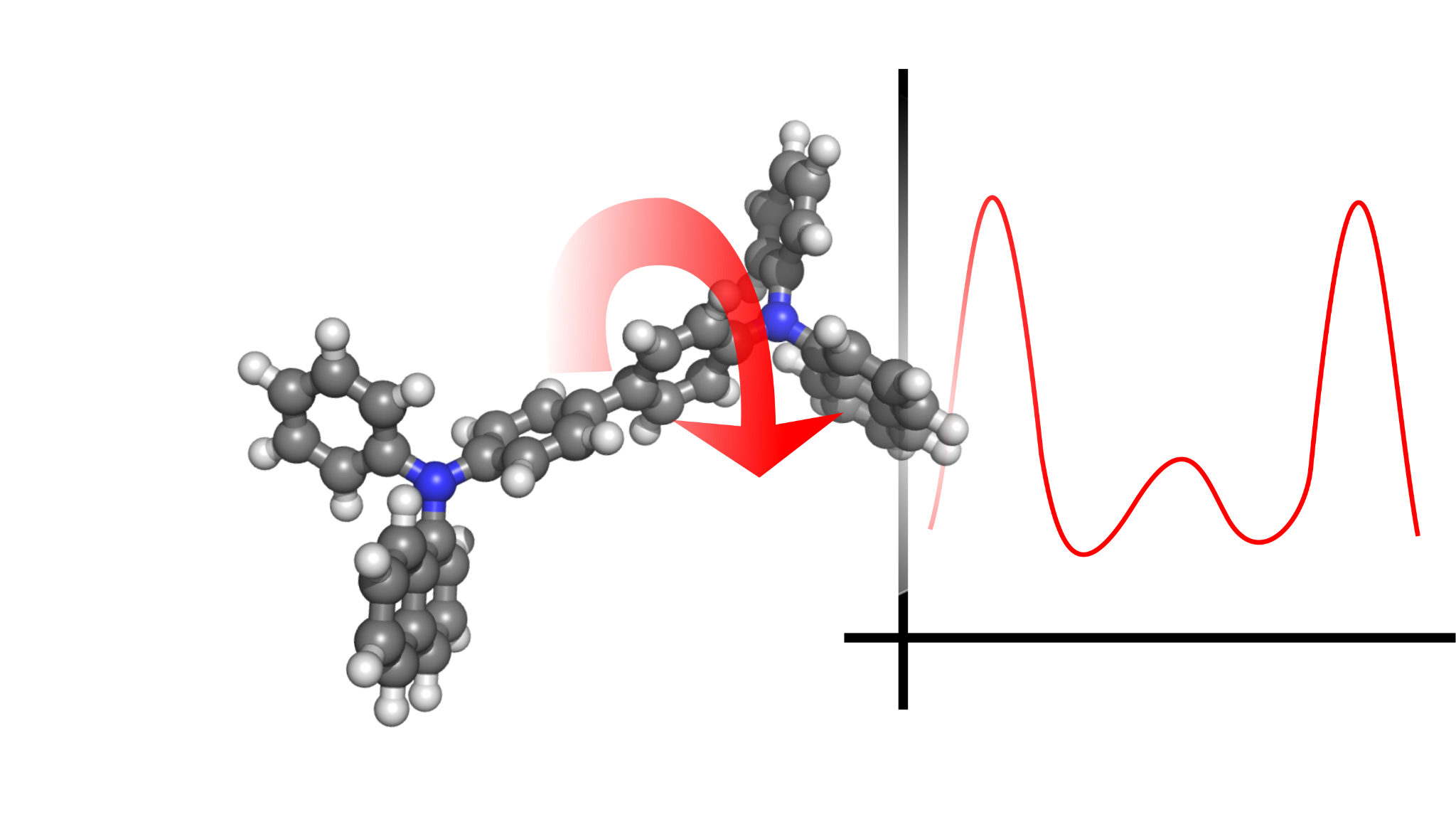
|
|
Simulation of the physical
vapor deposition (PVD)
to obtain atomistic morphology
|

|
Implemented Scientific Methods
The following scientific methods are implemented in the Nanomatch Morphology Calculator:
Molecular Structure Optimization
obabel, xtb, and Density Functional Theory (DFT) are used to generate initial 3D conformers, pre-optimize and optimize the geometry, and compute partial charges via an electrostatic potential (ESP) fit for single molecules in a vacuum. DFT optimization is performed with def2-TZVP/B3LYP level of theory.
Morphology Generation
The DEPOSIT protocol [1] simulates physical vapor deposition to generate thin-film morphologies with atomistic resolution. This involves Monte Carlo (MC) based basin hopping with simulated annealing (SA) to model intermolecular interactions during deposition. In total, 1000 molecules are deposited into a box with a base size of 100x100 Å.
Output
Displayed Results
The data below will be displayed as the workflow ends (backend name: result.yml):
ZUOUZKKEUPVFJK-UHFFFAOYSA-N:
HOMO:
value: -6.304540838835274
LUMO:
value: -0.9858224534777202
dipole:
results:
dipole_vector:
- -1.3524802844422331e-05
- 3.1223022592016277e-06
- 1.662349335263646e-05
value: 2.1656629345848317e-05
morphology:
results:
average_neighbors:
unit: Angstrom
value: 17.6
mass_density:
std: 0.01
unit: g/cm3
value: 1.14
molecular_volume:
unit: nm3
value: 0.23
number_density:
std: 9.9e+19
unit: 1/cm3
value: 4.36e+21
rdf_first_peak:
unit: Angstrom
value: 4.921630094043887
value: 'file: structure.cml'
The table below explains each parameter, its meaning, units, and other relevant information (field “value” is occasionally omitted).
Parameter |
Description |
Units |
Value |
Additional Information |
|---|---|---|---|---|
HOMO |
Highest Occupied Molecular Orbital |
eV |
-6.304540838835274 |
HOMO |
LUMO |
Lowest Unoccupied Molecular Orbital |
eV |
-0.9858224534777202 |
LUMO |
dipole_vector |
Components of the dipole moment vector |
Debye (D) |
(-1.3524802844422331e-05, 3.1223022592016277e-06, 1.662349335263646e-05) |
Vector representation of the dipole moment |
dipole |
Magnitude of the dipole moment |
Debye (D) |
2.1656629345848317e-05 |
Magnitude of the dipole moment |
average_neighbors |
Average distance to neighboring molecules |
Å |
17.6 |
Average distance to neighboring molecules |
mass_density |
Mass per unit volume |
g/cm³ |
1.14 |
Standard deviation (std): 0.01 (example value) |
molecular_volume |
Volume occupied by a single molecule |
nm³ |
0.23 |
|
number_density |
Number of molecules per unit volume |
1/cm³ |
4.36e+21 |
Standard deviation (std): 9.9e+19 (example value) |
rdf_first_peak |
Position of the first peak in the radial distribution function |
Å |
4.921630094043887 |
Indicates the most probable intermolecular distance |
Files
In addition to parsed output, the following files are available upon the workflow completion:
No. |
File |
Description |
Example |
|---|---|---|---|
1 |
output_molecule.mol2 |
Molecule output file in MOL2
format.
|
|
2 |
summary_RDF.png |
Radial distribution function
(RDF).
|
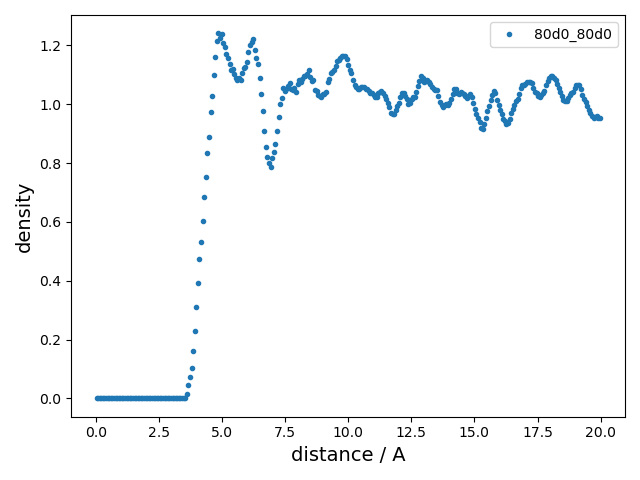
|
3 |
structure.cml |
Molecular structure in
CML format.
|
|
4 |
visualization_2D
_and_3D.png
|
2D and 3D visualizations
of the molecules
(center of geometries)
|
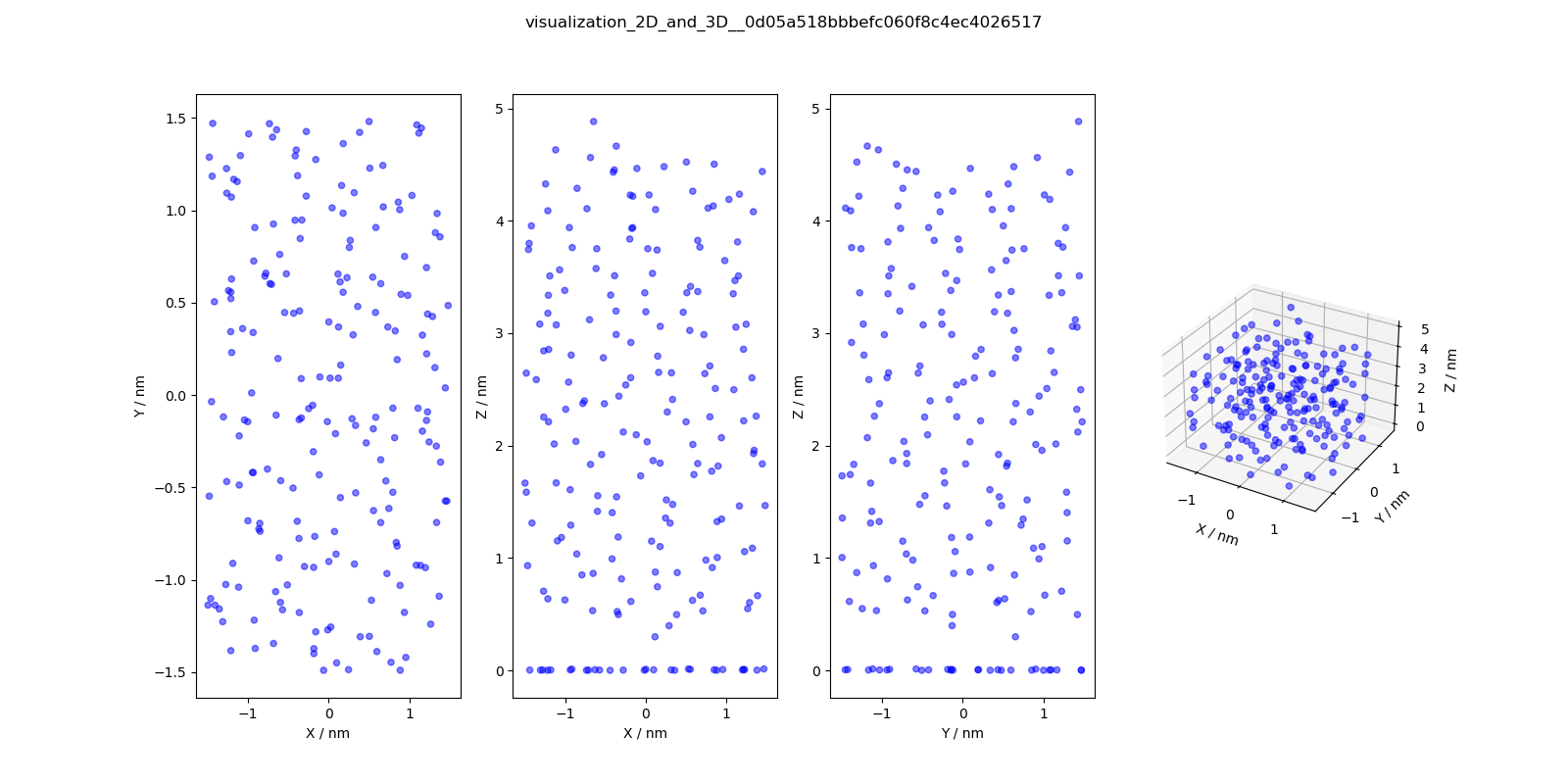
|